Overview of BATL
BATL ( Bayesian annotations for targeted lipidomics ) is a Gaussian naïve Bayes classifier for targeted lipidomics. BATL annotates peak identities according to user-specified peak features such as retention, intensity, and/or shape.
BATL calculates its statistical model based on training datasets with user assigned verified lipid identities to picked peaks. It then leverages that model to assign the most likely identities to peaks in new datasets lacking structural verification.
Training data for BATL should be produced following the 6-step process outlined below resulting in a lipid identity-labelled peak file used by BATL to train its statistical model. Example files can be downloaded from the Sample Data tab.
NOTE: Every time you wish to upload a new training set, you must reload https://complimet.ca/batl/ and refresh your browser.
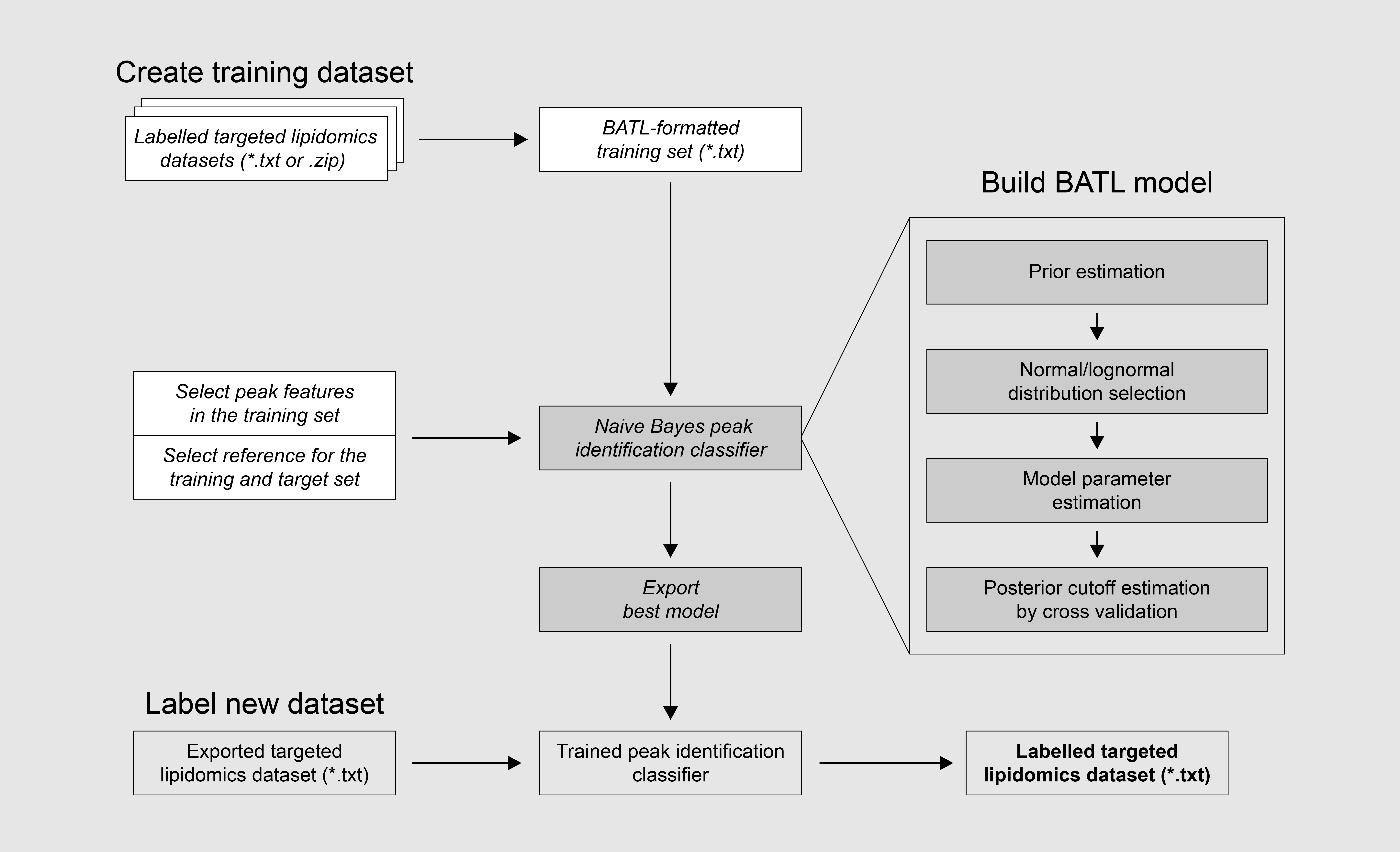
How to use BATL
Go to Analzye in the SideBar: Three tabs will be available
Tab 1 – Create Training Dataset
Prepare your labelled lipidomic datasets
Before using BATL online: Prepare your labelled lipidomic dataset(s) following this 6 step process:
-
In Sciex MultiQuant software, open a qsession for a dataset that you have fully analyzed.
If using other peak-picking softwares, please use our sample data files as a format template.
Note your final training datasets should contain at least 24 unique samples. You will be able to upload multiple .qsession files as a zip file and combine them into your training dataset following the instructions below.
-
Export the .qsession analysis. Ensure you have selected and exported all columns as a tab-delimited text (.txt) formatted file.
- File > Export > Results Table…
- Under Format > choose MultiQuant
- Under Columns > choose Export all columns
- Under Rows > choose Export all rows
- Name your file export and save it.
-
Lipid identities you wish to use to annotate each peak in your training datasets have to be verified prior to upload. BATL will annotate all subsequent datasets according to these identities. This identifier can be any standardized character string used by your laboratory to annotate lipid identities
-
Lipid identities you wish to use to annotate each peak in your training datasets have to be verified prior to upload. BATL will annotate all subsequent datasets according to these identities. This identifier can be any standardized character string used by your laboratory to annotate lipid identities.
-
Open the .txt quantification file, add a column called Lipid_identifier (case-sensitive) to the very end of each training dataset. This will be column CJ. (If it is not CJ then you have not exported all columns.
-
Save these file(s) as tab-delimited text (.txt) files. The features associated with these identities will form the base library that will be used to generate your statistical model.
Upload your BATL-formatted training datasets
-
Your quantification file is now labelled, is in BATL-compatible format, and can be used as your Training Dataset in Tab 1. Upload one BATL-formatted file (.txt), or multiple BATL-formatted files (zipped) to create your Training Dataset.
-
Specify the desired peak features to train the BATL model by selecting the appropriate check boxes.
-
If your peak features require normalization to a reference analyte that analyte must be present in all samples and must have the same component name in all training dataset files. Select that standard from the drop down menu. (Only analytes that have the same component name in all of your training dataset files and are present in every sample will be available for selection).
-
Your training dataset can now be built and downloaded for your reference. Proceed to Tab 2.
Tab 2. Build BATL Model
Build your statistical model
If you have not yet created your training set, you will not see the option to build your BATL model. Please return to tab 1 and create your training set.
-
Click on 'Click here to build your model'. Depending on the size of your training dataset, this may take up to 30 min. Enter your email to receive your file once completed.
-
Once your model is built, you can download it for your reference. Proceed to tab 3.
Tab 3. Label new dataset
Annotate new qsession file exports.
-
Upload your qsession export. File must be .txt and you must include all columns as above. If your model requires normalized features, the datasets you are annotating must have the same reference analyte in all samples and this reference analyte must have the same component name as in your training dataset.
-
Select the two letter Lipid Maps category that corresponds to your dataset. This selection is used to annotate potential in-source artifacts (i.e., dehydrations, isotopologues, etc). If you choose SP this will enable the assignment of dehydration/deglycosylation/dimer artifacts and isotopes that are relevant only to sphingolipids. Currently, only isotopologue annotations are supported for the remaining categories.
-
Click to annotate your quantification files. This may take up to 10 minutes depending on dataset size and bandwidth.
-
Annotated files can be downloaded.
Example training datasets available for download. All labelled lipid identities were confirmed by high performance liquid chromatography, selected reaction monitoring-information dependent acquisition-enhanced product ion scan electrospray ionization tandem mass spectrometry (LC-IDA-EPI-ESI-MS/MS) experiments. Lipid identities are indicated as barcodes used by the India Taylor Lipidomic Research Platform, University of Ottawa.
Training .qsession exports with Lipid_identifier column added (Use these datasets in Analyze Tab 1 to create your training sets).
-
PlasmaGlycerophosphocholinesvA.txt : 12 human plasma samples positive or negative for SARS-CoV-2.
-
PlasmaGlycerophosphocholinesvB.txt : 12 additional human plasma samples positive or negative for SARS-CoV-2.
In these training datasets, lipid identities were confirmed by LC-IDA-EPI-ESI-MS/MS using SRM as the survey scan. Analyses of EPI spectra were further validated by analyzing standards available from Avanti Polar Lipids. Users are advised to download both files and .zip to ensure your sample training set includes 24 samples.
Test .qsession exports for annotation (Use these datasets in Analyze Tab 3 as test sets to explore lipid annotation)
-
PlasmaGlycerophosphocholinesv_test.txt : 12 human plasma samples positive or negative for SARS-CoV-2.
Troubleshooting BATL
The most common reason for BATL failing to generate a model or annotate peaks is related to how your data (training and test sets) have been formatted. Please review common issues below and should BATL not perform as expected please email ldomic@uottawa.ca for assistance. In your help request, please provide your training datasets and test dataset and a description of how BATL is not performing to expectation. We will reproduce the problem and provide you with a solution.
1. My training datasets generated using Agilent or Waters acquisition software will not load.
The current BATL version supports SCIEX MultiQuant analyses or files converted to match our sample data. Future updates will expand capacity to include direct input of file formats from other targeted lipidomic software packages. Please reach out to us at ldomic@uottawa.ca and we will notify you when the next release becomes available.
2. My high resolution high mass accuracy training and test datasets will not load.
BATL was designed to verify and annotate peaks acquired through a targeted lipidomic workflow. This includes mass spectrometry acquisitions that were generated from multiple-reaction monitoring (MRM), scheduled MRM (sMRM), or selected reaction monitoring (SRM) experiments as well as information-dependent acquisitions (IDA). Thus, BATL requires mass information for both product and precursor ion monitored using triple quadrupole or QTRAP mass spectrometers.
3. My training datasets generated from .qsession analyses exported from SCIEX MultiQuant quantitation software will not load.
Ensure you have exported all columns (and not simply columns you routinely use). Also ensure you have added the required Lipid_identifier (case sensitive) column to your training files. If you have exported all columns correctly, you will be adding Lipid_identifier to column CJ.
Export the .qsession analysis. Ensure you have selected and exported all columns as tab-delimited text (.txt) formatted file.
- File > Export > Results Table…
- Under Format > choose MultiQuant
- Under Columns > choose Export all columns
- Under Rows > choose Export all rows
- Name your file export and save it.
Open your file with any text/spreadsheeting editing software. Go to the last column of your dataset (column CJ if using Microsoft Excel). Add a new column labelled Lipid_identifier and add your validated identity.
4. I have chosen to model features that require normalization but no reference analyte appears in the pull down menu. I cannot build my BATL model.
The reference analyte must be in all samples in each qsession export. If you are combining multiple qsession export training datasets, at least one reference analyte must be present in all files (and all samples). The Component Name of this reference analyte must be identical in all training dataset files (case sensitive).
5. I successfully generate a BATL model but when I upload the file(s) to be annotated, BATL returns the error 'Some peaks have no matching reference standard for feature normalization. Please ensure that <name of reference analyte> is detected in each of your samples you would like to annotate'.
The datasets you are annotating (and each sample in these datasets) must contain the same reference analyte (with the identical Component Name, case sensitive) as the one you used to normalize features in your BATL training datasets and model.
6. The BATL annotation indicates one of my species is potentially an in-source dehydration but I am annotating phospholipids. This is chemically impossible.
You likely indicated on Tab 3 that the 2-letter code of your analytes was SP (Sphingolipids). This dictates what in-source artifacts or isotopologues are interrogated. If your datasets were a mix of lipid categories then you should chose SP as the default to have all lipid artifacts assessed but you must curate the potential list of artifacts/isotopologues to ensure chemical probability. BATL provides this information to enable you to make informed decisions about the accuracy of your peak picking.
Contact us
Cite the use of BATL in a publication
Chitpin JG, Surendra A, Nguyen TT, Taylor GP, Xu H, Alecu I, Ortega R, Tomlinson JJ, Crawley AM, McGuinty M, Schlossmacher MG, Saunders-Pullman R, Cuperlovic-Culf M, Bennett SAL, Perkins TJ (2021) BATL: Bayesian annotations for targeted lipidomics. Bioinformatics 38:1593-1599, doi.org/10.1093/bioinformatics/btab854
Public Server
BATL: https://complimet.ca/batl/
Software License
BATL is free software. You can redistribute it and/or modify it under the terms of the GNU General Public License v3 (or later versions) as published by the Free Software Foundation. As per the GNU General Public License, BATL is distributed as a bioinformatic lipidomic tool to assist users WITHOUT ANY WARRANTY and without any implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. All limitations of warranty are indicated in the GNU General Public License.